Recent Publications
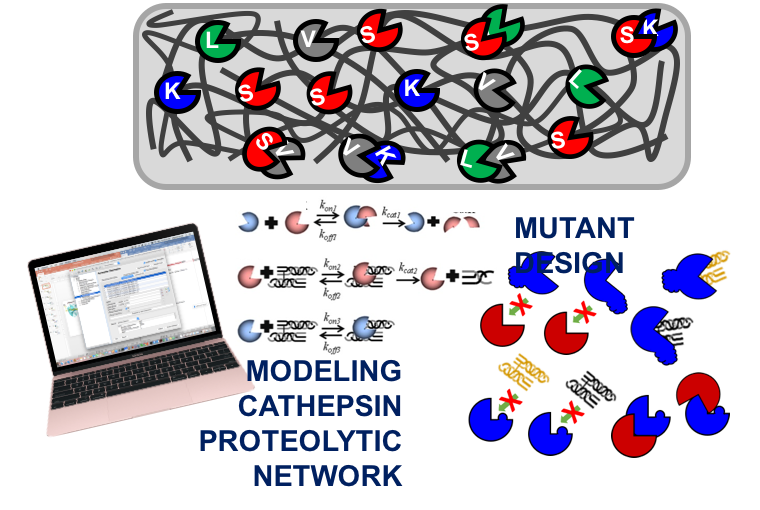
Reassessing enzyme kinetics: Considering protease-as-substrate interactions in proteolytic networks
Proteases are enzymes that hydrolyze other proteins, including other proteases, which challenges assumptions of enzyme inertness in chemical reactions, and alters predicted protease−substrate concentrations using established mass action frameworks. Cysteine cathepsins are powerful proteases involved in numerous diseases by cleaving substrates, but they also hydrolyze each other, requiring inclusion of as yet undefined, protease-as-substrate dynamics. Here, we used experimental and computational models to improve predictions of the concentrations of multiple species and intermediates generated during substrate degradation in multiprotease systems by including protease-on-protease reactions of autodigestion, inactivation, cannibalism, and distraction in proteolytic networks. This was made available online for others to test perturbations and predict shifts in proteolytic network reactions and system dynamics (https://plattlab.shinyapps.io/catKLS/).
PNAS
Re: Simulation analysis for tumor radiotherapy based on three-component mathematical models.
Hong & Zhang’s “Simulation analysis for tumor radiotherapy based on three-component mathematical models” in the recent issue of JACMP addressed an important topic that deserves much attention, because mathematical predictions of treatment response may ultimately help personalize radiation dose and fractionation. Hong & Zhang conclude from model simulation that a three-compartment model impacts radiotherapy efficacy and outcome of simulated radiotherapy was affected by the proportion of quiescent tumor cells, radiation dose per fraction, and radiosensitivity. Although intuitive and well-supported by literature, we found selected discrepancies and inaccuracies with regards to these specific claims, in addition to several discrepancies in the mathematical formulation of their model, which we describe here in a Letter to the Editor.
J Appl Clin Med Phys
Long-term cryopreservation and revival of tissue engineered skeletal muscle.
Tissue engineered skeletal muscle plays an important role not only in the field of regenerative medicine, but also in emerging areas such as soft robotics, organ-on-a-chip disease models, and drug testing. However, further expansion of the applications of tissue engineered skeletal muscle models requires a suitable method for their long-term storage and shipment. Cryopreservation has long been the standard for cell storage, but the freezing of 3D tissues is accompanied by many complications due to heat and mass transfer limitations. In this study, we used a tissue engineered skeletal muscle bioactuator as a model to characterize the effects of freezing on muscle viability, gene expression, myotube structure, and force generation. We optimized the protocol for cryopreservation by comparing outcomes when tissue was frozen undifferentiated and differentiated. Our optimized protocol, in which skeletal muscle was frozen undifferentiated, not only maintained cell viability, but led to a 3-fold increase in force production as compared to unfrozen muscle. Furthermore, we enhanced muscle lifetime through inhibition of cysteine proteases. The reported timeline for skeletal muscle tissue fabrication, freezing, revival, and long-term culture not only promotes a more streamlined fabrication process, but enables multi-site collaborative research efforts through the shipment of pre-formed skeletal muscle constructs.
Tissue Eng Part A
Leveraging single cell RNA sequencing experiments to model intra-tumor heterogeneity
PURPOSE: Many cancers can be treated with targeted. Almost inevitably, tumors develop resistance to targeted therapy, either from preexistence or by evolving new genotypes and traits. Intra-tumor heterogeneity serves as a reservoir for resistance, which often occurs due to selection of minor cellular sub-clones. On the level of gene expression, the { extquoteright}clonal{ extquoteright} heterogeneity can only be revealed by high-dimensional single cell methods. We propose to use a general diversity index (GDI) to quantify heterogeneity on multiple scales and relate it to disease evolution. METHODS: We focused on individual patient samples probed with single cell RNA sequencing to describe heterogeneity. We developed a pipeline to analyze single cell data, via sample normalization, clustering and mathematical interpretation using a generalized diversity measure, and exemplify the utility of this platform using single cell data. RESULTS: We focused on three sources of RNA sequencing data: two healthy bone marrow (BM) samples, two acute myeloid leukemia (AML) patients, each sampled before and after BM transplant (BMT), four samples of pre-sorted lineages, and six lung carcinoma patients with multi-region sampling. While healthy/normal samples scored low in diversity overall, GDI further quantified in which respect these samples differed. While a widely used Shannon diversity index sometimes reveals less differences, GDI exhibits differences in the number of potential key drivers or clonal richness. Comparing pre and post BMT AML samples did not reveal differences in heterogeneity, although they can be very different biologically. CONCLUSION: GDI can quantify cellular heterogeneity changes across a wide spectrum, even when standard measures, such as the Shannon index, do not. Our approach offers wide applications to quantify heterogeneity across samples and conditions.
biorxiv
Computational predictions of cysteine cathepsin-mediated fibrinogen proteolysis
Fibrin clot formation is a proteolytic cascade of events with thrombin and plasmin identified as the main proteases cleaving fibrinogen precursor, and the fibrin polymer, respectively. Other proteases may be involved directly in fibrin(ogen) cleavage, clot formation, and resolution, or in the degradation of fibrin-based scaffolds emerging as useful tools for tissue engineered constructs. Here, cysteine cathepsins are investigated for their putative ability to hydrolyze fibrinogen, since they are potent proteases, first identified in lysosomal protein degradation and known to participate in extracellular proteolysis. To further explore this, we used two independent computational technqiues, molecular docking and bioinformatics sequence analysis (PACMANS), to predict potential binding interactions and sites of hydrolysis between cathepsins K, L, and S and fibrinogen. By comparing the results from these two objective, computational methods, it was determined that cathepsins K, L, and S do bind and cleave fibrinogen α, β, and γ chains at similar and unique sites. These differences were visualized experimentally by the unique cleaved fibrinogen banding patterns after incubation with each of the cathepsins, separately. In conclusion, human cysteine cathepsins K, L, and S are a new class of proteases that should be considered during fibrin(ogen) degradation studies both for disease processes where coagulation is a concern, and also in the implementation and design of bioengineered systems.
Protein Science

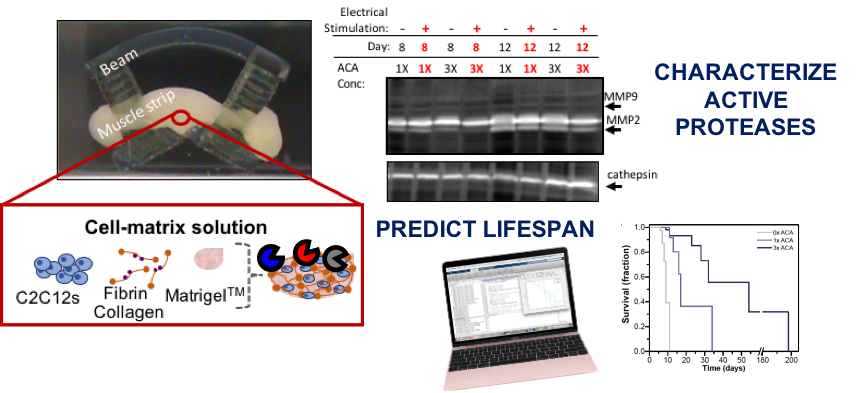
Investigating the Life Expectancy and Proteolytic Degradation of Engineered Skeletal Muscle Biological Machines
A combination of techniques from 3D printing, tissue engineering, and biomaterials has yielded a new class of engineered biological robots that could be reliably controlled via applied signals. These machines are powered by a muscle strip composed of differentiated skeletal myofibers in a matrix of natural proteins, including fibrin, that provide physical support and cues to the cells as an engineered basement membrane. However, maintaining consistent results becomes challenging when sustaining a living system in vitro. Skeletal muscle must be preserved in a differentiated state, and the system is subject to degradation by proteolytic enzymes that can break down its mechanical integrity. Here we examine the life expectancy, breakdown, and device failure of engineered skeletal muscle bio-bots as a result of degradation by three proteases: plasmin, cathepsin L, and matrix metalloproteinases (MMP-2 and MMP-9). We also demonstrate the use of gelatin zymography to determine the effects of differentiation and inhibitor concentration on cysteine protease expression. With this knowledge, we are poised to design the next generation of complex biological machines with controllable function, specific life expectancy, and greater consistency. These results could also prove useful for the study of disease-specific models, treatments of myopathies, and other tissue engineering applications.
Scientific Reports
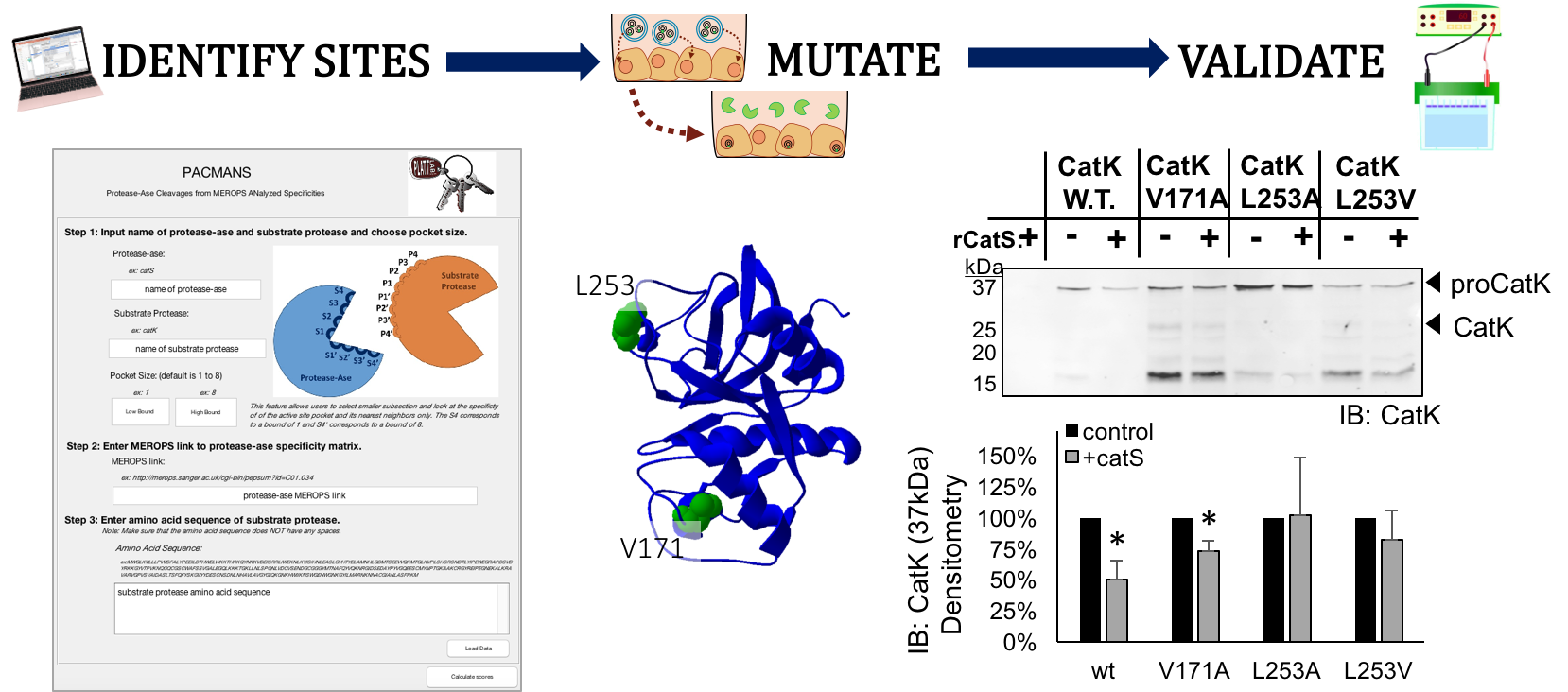
PACMANS: A Bioinformatically Informed Algorithm to Predict, Design, and Disrupt Protease-on-Protease Hydrolysis
To explore potential protease-on-protease interactions in silico, a program was developed for users to input two proteases: 1) the protease-ase that hydrolyzes 2) the substrate, protease. This program identifies putative sites on the substrate protease highly susceptible to cleavage by the protease-ase, using a sliding window approach that scores amino acid sequences by their preference in the protease-ase active site, culled from MEROPS database. We call this PACMANS, Protease-Ase Cleavage from MEROPS Analyzed Specificities, and test and validate this algorithm with cathepsins S and K.
Protein Science Volume 26, Issue 4 Pages 880-890